Eine KI die Proteine versteht: Die CAFA Challenge
Programmiersprache: Python
Genutzte Software: Tensorflow, Keras, scikit-learn, Pandas, InterproScan, Diamond, BioPython, NetworkX
Keywords: Machine Learning, Neuronale Netze, RNN, NLP, LSTM, Word Embeddings, Random Forests, Bioinformatik, UniProt, Interpro, FASTA, Gene Ontologies, Graphentheorie, Kaggle
Inhaltsverzeichnis:
- Eine KI die Proteine versteht: Die CAFA Challenge
- Das Setup
- Daten Analyse der CAFA Challenge
- Ansatz: Wie kommt man vom Protein zu den GO Terms?
- Protein KI mit CNNs und ComboLoss
- Die Protein KI mit neuem Input: K-Mers
- t-SNE: Wie gut sind die k-Mer Features?
- Protein KI ohne Neuronales Netz? 1-vs-Rest
- Domains, Motifs und Superfamilies: Das InterPro Model
- Mehr als nur ein Input für die Protein-KI?
- Proteine in Zahlen übersetzen: Embeddings
Das Setup
Am 18. April 2023 wurde die CAFA Challenge auf Kaggle veröffentlicht. Die Aufgabe war, aus einer Proteinsequenz dessen Funktion zu erkennen.
Aber warum ist das überhaupt wichtig? Proteine sind die Arbeiter in jedem Organismus. Sämtliche biologischen Vorgänge werden durch Proteine ermöglicht und daher gibt es viele Zehntausend verschiedene Arten.
Proteine entdecken und deren Bestandteile herauszufinden ist einfacher als die genaue Funktion zu bestimmen.
Generell ist die Einteilung von Funktionen schwierig, wenn man bedenkt, wie viele verschiedene biologische Prozesse es gibt. Und dafür wurde die Gene Ontology (GO) eingeführt:
Das sind ungefähr 43.000 Terms in einer baumartigen Struktur. Diese Struktur kann als Graph behandelt werden und so hat z.B. ein bestimmter Term mehrere Vor- und Nachfahren.
An der “obersten” Stelle dieser Gene Ontology (GO) gibt es drei grundlegende Sub-Ontologies:
- Cellular Component: Wo genau sich etwas in der Zelle befindet.
- Molecular Function: Was für eine physikalische oder chemische Funktion es ist.
- Biological Process: In welchem biologischen Prozess es involviert ist.
Unter diesen drei Sub-Ontologies gibt es nun viele Möglichkeiten. Zwei Beispiele:
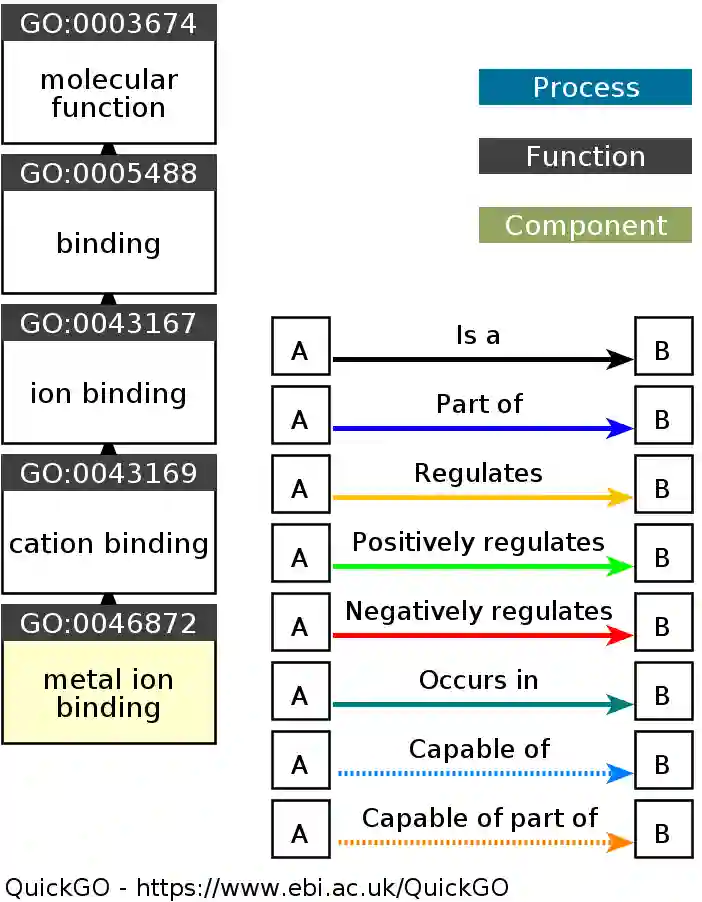
Dieses einfache Beispiel einer Metall-Ionen-Bindung enthält 5 GOs. Die Metall-Ionen Bindung ist eine Kationen Bindung (GO:0043169), was eine Ionenbindung (GO:0043167) ist, was eine Bindung (GO:0005488) und letzen Endes eine molekulare Funktion ist.
Deutlich schwieriger wird es, je tiefer man sich in den Graph bewegt:
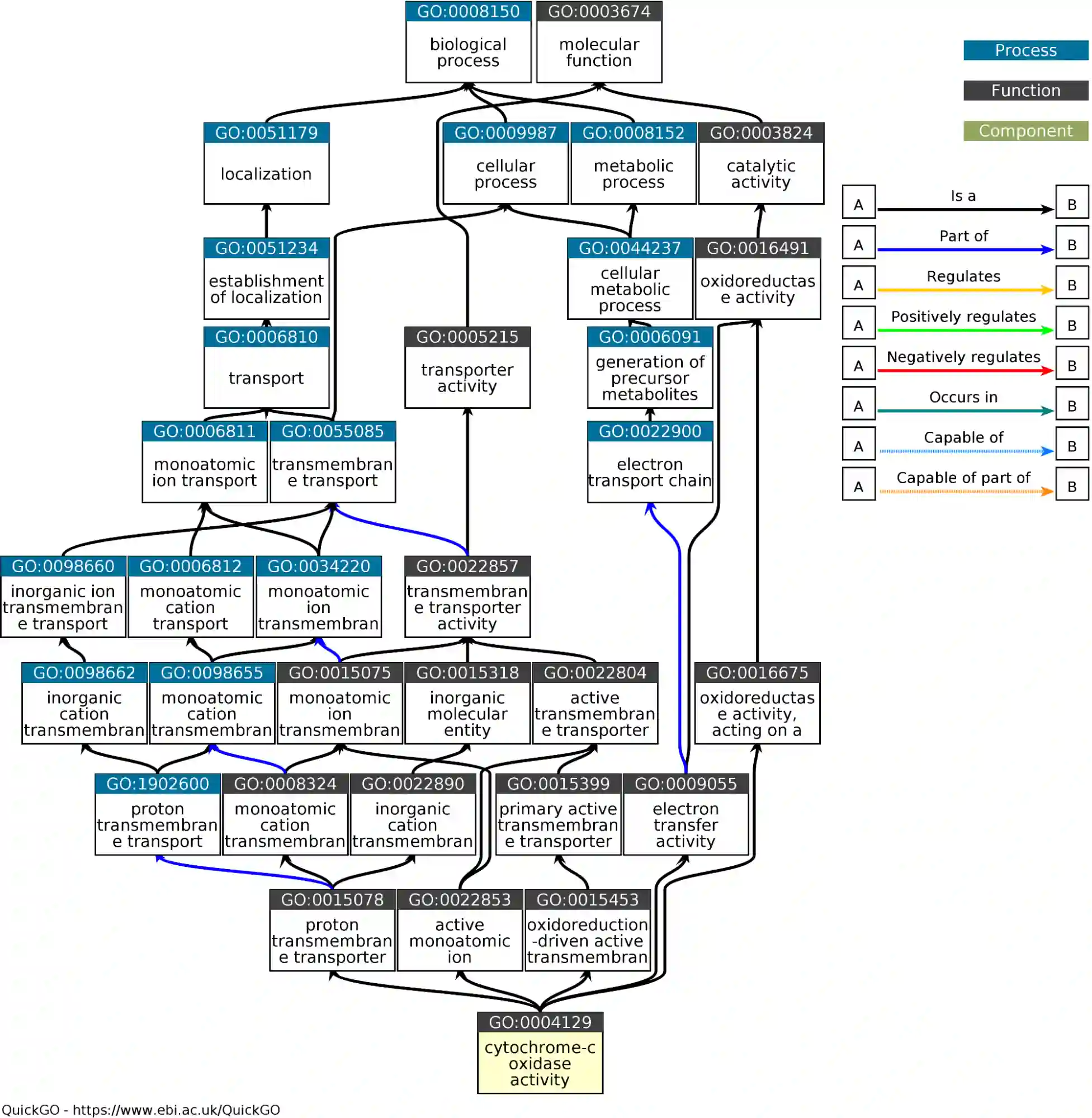
Die cytochrome-c oxidase activity ist sowohl Teil eines biologischen Prozesses als auch eine molekulare Funktion mit vielen Unter-GOs.
Die “True Path”-Rule besagt, dass alle GOs, die zwischen einer zugewiesener GO auf dem Weg zum Ursprung liegen, auch Teil dieser Zuweisung sind.
In unserem Beispiel hier hätte also ein Protein, das man mit cytochrome-c oxidase activity in Verbindung bringt, auch alle anderen GOs in dem Graph zugeordnet.
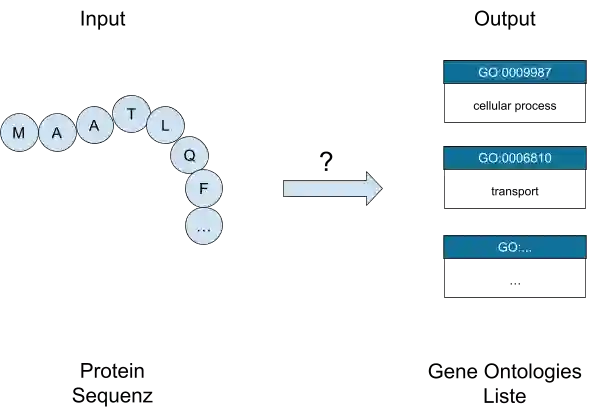
Nun wissen wir, was der Output sein soll. Die KI soll einem Protein möglichst alle Gene Ontologies zuordnen.
Der Input ist dabei recht einfach: Das Protein als Abfolge von Aminosäuren (Primärstruktur). Also eine Liste von Buchstaben, wobei ein Buchstabe einer Aminosäure entspricht.
Unser Vokabular hat damit 22 möglichen Aminosäuren (AS) die es in der Biologie gibt + 3 für unbekannte/unbestimmte Aminosäuren:
- A : Aspartam
- R: Arginin
- …
- B: Asparaginsäure oder Asparagin
- X: Unbekannte Aminosäure
Ein Beispiel für ein Protein im FASTA Format:
>Q6P0A1 sp|Q6P0A1|F180B_HUMAN Protein FAM180B OS=Homo sapiens OX=9606 GN=FAM180B PE=1 SV=3
MAATLQFLVCLVVAICLLSGVTTTQPHAGQPMDSTSVGGGLQEPEAPEVMFELLWAGLEL
DVMGQLHIQDEELASTHPGRRLRLLLQHHVPSDLEGTEQWLQQLQDLRKGPPLSTWDFEH
LLLTGLSCVYRLHAASEAEERGRWAQVFALLAQETLWDLCKGFCPQDRPPSLGSWASILD
Die Problemstellung ist hier nochmal in einem Diagramm dargestellt
Daten Analyse der CAFA Challenge
Ein paar Eckdaten zu der CAFA Challenge :
- Es wurden ~140.000 Sequenzen und deren GO Zuordnungen zum Training bereitgestellt
- Ebenfalls ~140.000 Sequenzen sind im Test-Datensatz (mit unbekannter Funktion) und sollen vorhergesagt werden
- Insgesamt gibt es ~5 Mio. Zuordnungen
- Es werden ca. 31.000 GO Terms verwendet
- 21.000 unter “biological process”
- 7.000 unter “molecular function”
- 3.000 unter “cellular component”
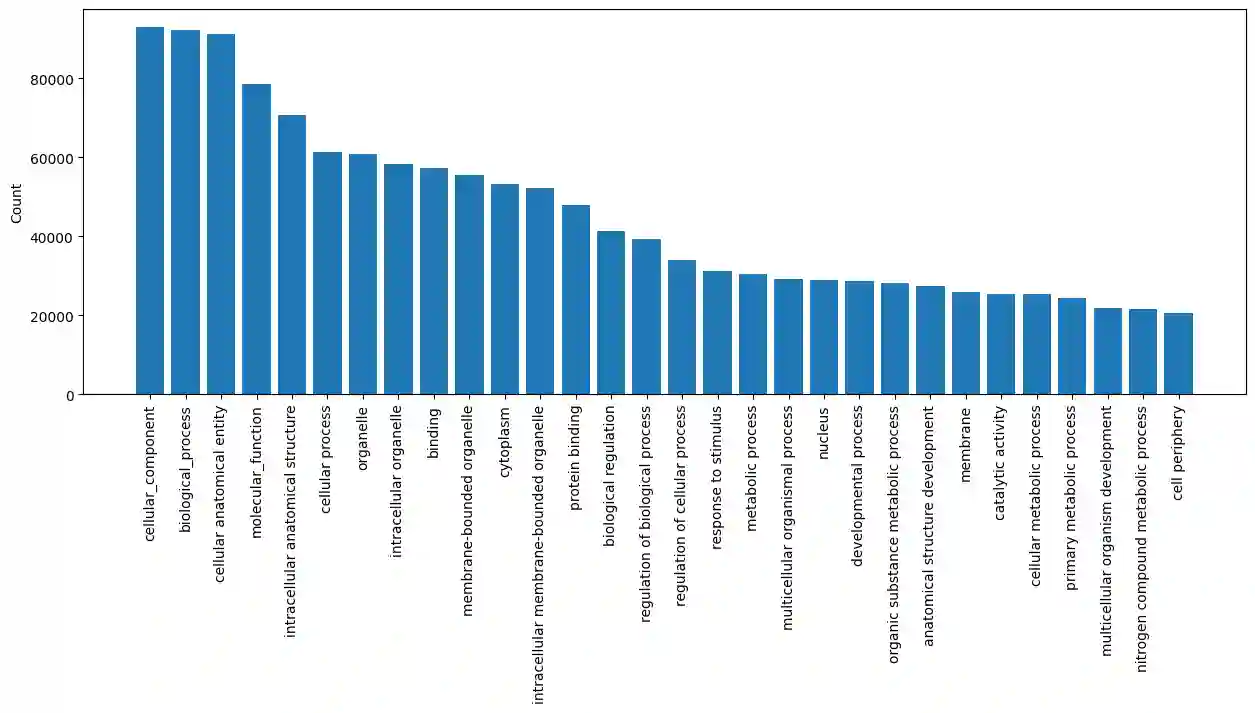
Hier ist die Zuordnung der 30 häufigsten Terms zu sehen. Die Sub-Ontologies sind natürlich ganz vorne dabei (cellular component, biological process und molecular function). Aus diesem Diagramm erkennt man auch, dass nicht alle 140k Sequenzen in allen Sub-Ontologies vertreten sind.
Am anderen Ende von diesem Histogramm gibt es aber auch Terms die nur ein einziges mal zugeordnet worden sind.
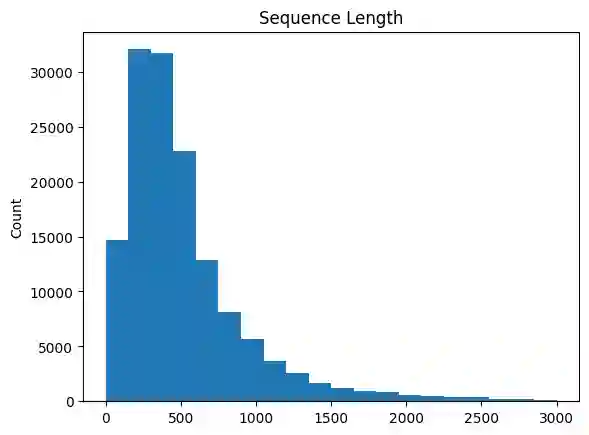
Die Länge der Sequenzen beträgt meist um die 250-1000 Aminosäuren. Es gibt allerdings einige wenige Ausreißer mit bis zu 35.000 Aminosäuren.
Ansatz: Wie kommt man vom Protein zu den GO Terms?
In dem Übersichtsdiagramm oben sieht das Ganze ja sehr einfach aus: Wir haben Sequenzen aus 25 Buchstaben und das soll in die GO Terms übersetzt werden. Für eine KI sollte das ja wohl kein Problem sein, oder?
Dabei gibt es aber ein paar Dinge zu beachten.
Beim Input:
- Machine Learning Algorithmen mögen
keine
Buchstaben.
ML basiert immer auf Zahlen und mathematischen Operationen.Buchstaben müssen also immer sinnvoll zu Zahlen übersetzt werden. - Sequenzen sind variabel.
Die Länge der Sequenzen variiert von ein paar wenigen AS zu 35.000. Und mit der Länge variiert auch die Komplexität der Aufgabe.
Beim Output:
- Die GO Terms sind hierarchisch.
Wenn ein Term tief im GO Graph vorhergesagt wird, gilt das auch für alle Vorfahren. - Die Aufgabe ist eine Multi-Label
Klassifizierung.
Häufig gibt es Klassifizierungen, bei denen ein Sample genau einer Klasse zugeordnet wird (z.B. das Bild einer Katze wird der Klasse “Katze” zugeordnet). Hier haben wir es aber mit mehreren Klassen pro Instanz zu tun. Ein Protein wird vielen verschiedenen GO-Terms zugeordnet.
Für jeden dieser Punkte gibt es natürlich Lösungen:
Punkt 1: Um Klassen oder Buchstaben in Zahlen zu übersetzen, ist der
einfachste
Ansatz die
simple Zuweisung von Zahlen. Jeder Buchstabe
bekommt eine Zahl
von 0 -
M zugewiesen, wobei M die Größe des Vokabulars ist.
Eine andere
Möglichkeit ist
das
One-Hot Encoding. Jeder Buchstabe wird in einen Vektor übersetzt. Dieser
hat die Länge N.
Der
Vektor ist 1 an der Stelle des entsprechenden Buchstabens und sonst 0.
Punkt 2: Mit RNNs (Recurrent Neural Networks) können Sequenzen beliebiger Länge verarbeitet werden.
Punkt 3: Ein neuronale Netz kann solche Zusammenhänge verstehen und vorhersagen
Punkt 4: Eine Multilabel-Klassifizierung macht die Aufgabe zwar deutlich schwieriger, aber ein neuronales Netz kann beliebig viele Output Werte liefern. Für unsere Zwecke wird das die Wahrscheinlichkeit für alle GO Terms sein.
Mein erster Ansatz eines neuronalen Netzes sieht dann so aus:
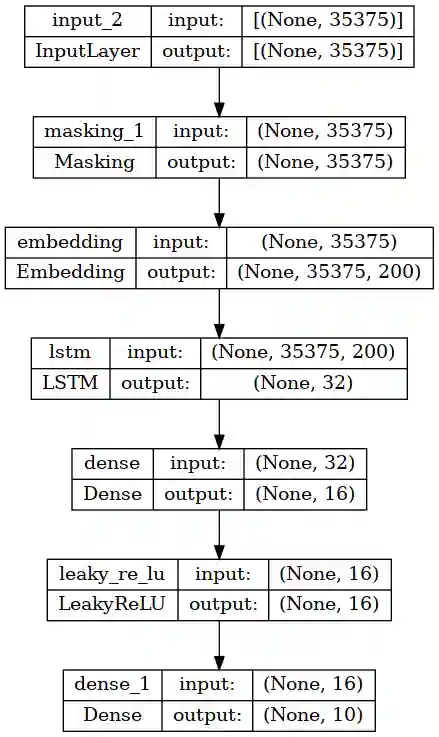
Zu Testzwecken habe ich hier nur die 10 häufigsten GO Terms als Output gewählt.
Mit diesem RNN werden die einzelnen Aminosäuren nacheinander vom Netzwerk verarbeitet und die Zwischenergebnisse immer wieder als Input genutzt.
Dieses Model lernt zwar schon, allerdings mit keiner wirklich guten Performance. Dafür benötigt es ein paar Upgrades:
Protein KI mit CNNs und ComboLoss
Immer wenn ich mit RNNs arbeite, taucht leider ein wohlbekanntes Problem auf: Vanishing Gradient. Das passiert meist dann, wenn die Sequenzen so lang sind, dass der Gradient (= der Anpassung Schritt des neuronalen Netzes an die Daten) so gering wird, dass das Netzwerk überhaupt nichts mehr lernt.
Ein komplett anderer Ansatz muss her: Convolutional Neural Networks (CNNs) sind sehr gut geeignet, um wichtige Features und Muster zu erkennen. Das funktioniert aber nur mit einer festen Input-Größe. Daher verwende ich hierfür folgendes Preprocessing der Sequenzen:
Aus der Datenanalyse ist bekannt, dass es nur sehr wenige Proteine mit mehr als 5000 Aminosäuren gibt. Daher werden alle Sequenzen auf eine Länge von 5000 gekürzt oder mit Padding gestreckt.
Damit hat der Input immer die genau gleiche Dimension von (5000,1). Das verwendete Model ist ein simples 1D-Convolution Model mit Dense Layers am Ende:
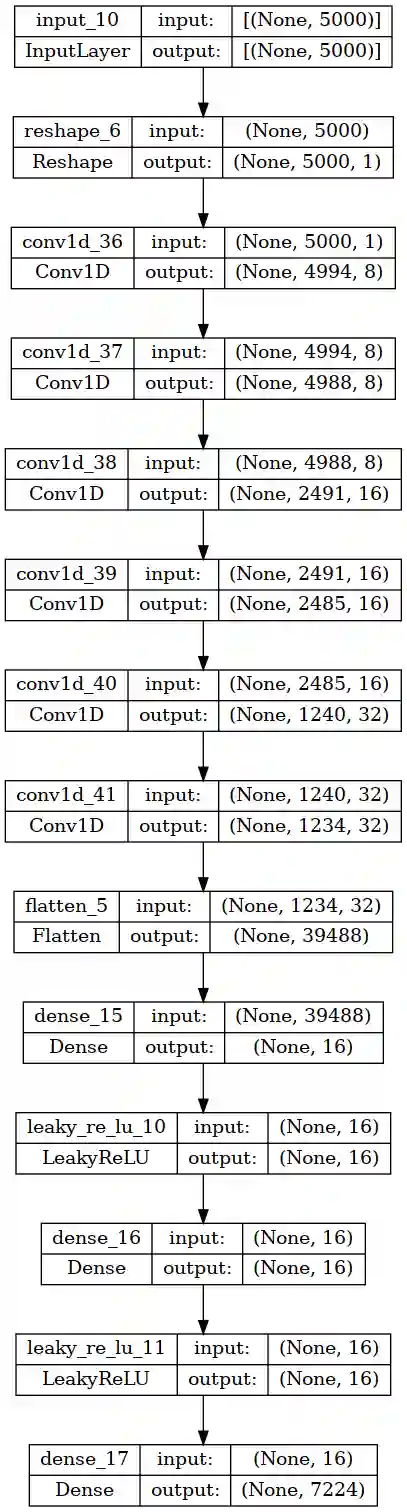
Jedes zweite Convolution Layer hat ein Stride-Parameter von 2, d.h. Der Input wird um den Faktor 2 runterskaliert und dabei wird die Filteranzahl um den Faktor 2 erhöht.
Das ist ein übliches Vorgehen bei CNNs, wie z.B. auch in der ResNet Architektur zu sehen.
Der Output sind die 7224 GO Terms der “molecular function” (MFO) Subontolgy.
Zusätzlich zu dieser neuen Architektur habe ich auch eine neue Loss Function eingeführt. Für gewöhnlich wird Cross Entropy dafür benutzt, allerdings ist das nicht immer optimal.
Gerade bei unterschiedlich häufigen Klassen (Class Imbalance) führt das oft zu einem ungleichmäßigen Lernen. Und genau das haben wir bei den GO Terms. Es gibt manche Terms die zu jedem Protein zugewiesen werden und andere, die so gut wie nie vorkommen.
Die Metrik, die für die CAFA-Challenge relevant ist, ist der F1-Score. Standardmäßig wird einfach nur die Accuracy angeschaut, allerdings sagt das nur wenig über die Performance des Classifiers aus. Für den F1-Score gibt es eine äquivalente Loss-Function, nämlich den Dice Loss.
Der ab hier verwendete Combo Loss kombiniert Cross Entropy und Dice Loss, um die Class Imbalance und False Positive/False Negative Imbalance möglichst gut auszugleichen.
Bei diesem Model für MFO ergibt sich auf dem Validierungs-Datensatz eine Accuracy von 99% und ein F1 Score von gerade mal 0,1.
Das ist ein sehr gutes Beispiel dafür, dass Accuracy keine wirklich sinnvolle Aussage über die Qualität eines Classifiers macht.
Ein guter Wert sollte F1>0,5 sein, d.h. die Daten, die das Model zur Verfügung hat (oder das Model selbst), reichen nicht aus, um die GO Terms zuverlässig vorauszusagen.
Die Protein KI mit neuem Input: K-Mers
Sequenzen mit variabler Länge sind einfach unhandlich. Ein neuronales Netz arbeitet am besten mit festen Features. Bei meinem Ansatz zuvor habe ich einfach alle Sequenzen auf 5000 gekürzt oder verlängert. Das funktioniert zwar, allerdings werden die Proteine damit eigentlich komplett verfälscht.
2021 haben Ali et al. den Spike2Vec Ansatz veröffentlicht. Das war noch mitten in der Corona-Pandemie. Es wurden viele Gensequenzen veröffentlicht und ihr Ansatz sollte helfen, dass Machine Learning Algorithmen effizient damit arbeiten können.
Der erste Schritt ist die Erstellung von k-Mers. D.h. man unterteilt die Sequenz in viele kleine Untersequenzen der Länge k. Der Faktor k wurde im Paper auf 3 gesetzt - was im biologischen Kontext auch Sinn macht: In der DNA wird jedes Basentriplett einer Aminosäure zugeordnet.
Danach erstellt man ein Histogramm aller möglichen
k-Mer.Das ist dann ein Vektor der Länge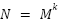 wobei M die
Größe des Vokabulars ist. Vokabular
heißt in unserem Fall die Menge aller Aminosäure und k=3, also
wobei M die
Größe des Vokabulars ist. Vokabular
heißt in unserem Fall die Menge aller Aminosäure und k=3, also 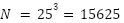 . An jeder
Stelle dieses Vektors wird
eingetragen,
wie oft
ein Triplett vorkommt.
. An jeder
Stelle dieses Vektors wird
eingetragen,
wie oft
ein Triplett vorkommt.
Und jetzt kommt der riesige Vorteil von diesem Ansatz: Für jede beliebige Proteinlänge ergibt sich ein Vektor der Länge 15625. Damit lässt sich jedes beliebige Model trainieren, also nicht nur neuronale Netze, sondern auch SVMs, Random Forests oder einfach nur Logistic Regressions.

Das damit trainierte Model ist ein Fully-Connected Neural Network mit Batch-Norm und Dropout.
Das Ergebnis: F1 = 0,26 auf dem Validierungs-Datensatz für CCP (cellular component).
Das ist ein deutlicher Sprung im Vergleich zu dem vorherigen Modell, und die k-Mer scheinen eine gute Möglichkeit zu sein, Proteine darzustellen.
Doch wie schon oben geschrieben: Das ist nur der halbe Weg, denn das Ziel sollte ein F1 Wert von über 0,5 sein.
Schauen wir uns also an, ob die k-Mer wirklich schon ausreichen, um einen guten Classifier daraus zu machen.
t-SNE: Wie gut sind die k-Mer Features?
t-distributed stochastic neighbor embedding. Hinter diesem unhandlichen Begriff steckt ein Verfahren, das hochdimensionale Features (für k-Mers ist die Dimension=15625) in 2D oder 3D transformiert.
Daten in 15.000 Dimensionen kann sich kein Mensch vorstellen. Daten in 2 Dimensionen schon - das kann nämlich einfach als Diagramm dargestellt werden.
Das Prinzip hinter t-SNE ist wie folgt: Es wird die Ähnlichkeit aller Samples berechnet und danach ein Mapping aller Samples in 2 oder 3 Dimensionen gemacht, sodass die Ähnlichkeiten gleich bleiben.
Das heißt, wenn man im t-SNE Plot mehrere Punkte auf einer Stelle sieht, sind diese Samples sehr ähnlich. Im besten Fall gibt es deutlich unterscheidbare Cluster von Punkten, denn dann ist der Datensatz auch durch einen Classifier gut unterscheidbar.
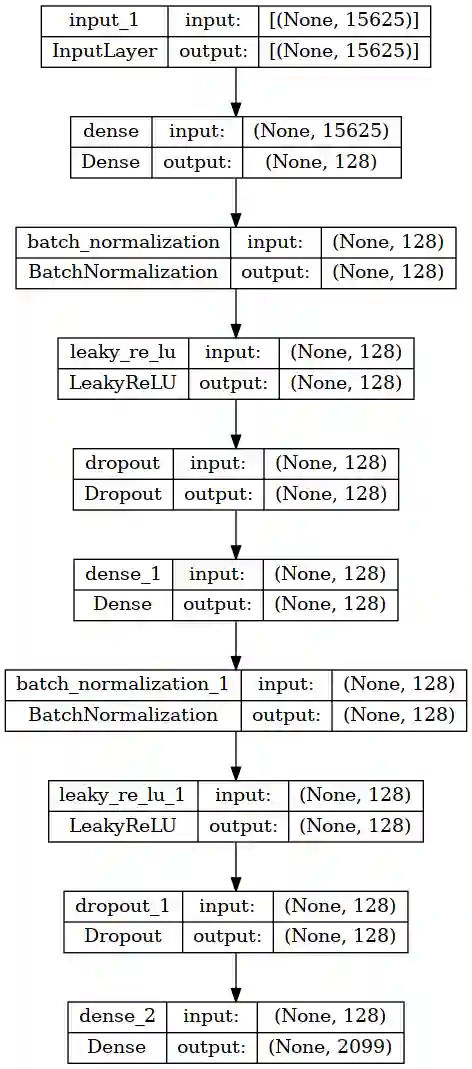
Für die k-Mer Features habe ich 6000 Samples zufällig aus dem Datensatz genommen und mit der t-SNE Transformation sieht das ganze so aus:
Jeder Punkt entspricht einem Sample. Man erkennt verschiedene Cluster von Punkten und einen Bogen im oberen linken Bereich.
D.h. man kann durch die k-Mers schon einige Klassen von Proteinen deutlich unterscheiden, aber man muss bedenken, dass der Output der GO Terms mehrere tausend Klassen enthält.
Protein KI ohne Neuronales Netz? 1-vs-Rest
Beim KI-Thema denkt man direkt an neuronale Netze. Aber das ist ja nur eine von vielen Methoden - wenn auch eine der erfolgreichsten. Es gibt viele weitere, die zum Teil sogar effizienter sind.
Z.B. Random Forests oder AdaBoost können als Classifier sehr gut performen, allerdings funktioniert das nicht mit mehreren verschiedenen Klassen. Dafür muss man die Algorithmen nochmal neu “verpacken”:
Ein 1-vs-Rest Ansatz sortiert die Daten zu Beginn neu und nutzt einen separaten Classifier für jede Klasse. Also hat man auch nur noch zwei Klassen: Die Gesuchte und den ganzen Rest.
Da auch in dem ursprünglichen Spike2Vec Paper ein Logistic Regression-Classifier (LogReg) sehr gut performt hat, war das auch hier mein Vorgehen.
Beim Testen hatte ich bereits Support Vector Machines, Random Forests und Logistic Regression verglichen, wobei meistens ähnliche Performances erzielt wurden. Der klare Vorteil der LogReg ist die Einfachheit: Es wird nur ein lineares Model an die Daten gefittet.
Mein Prozess habe ich mit sklearn implementiert und sieht folgendermaßen aus:
- Für eine GO einer Sub-Ontology, finde alle Sequenzen (bzw. deren k-Mers) denen diese GO zugeordnet ist. Das entspricht der positiven Klasse.
- Suche zufällig aus dem restlichen Datensatz Samples, denen diese GO nicht zugeordnet ist. Das entspricht der Restklasse. Die Anzahl wird so gewählt, dass beide Klassen gleich groß sind.
- Mache einen Train-validation-Split mit 25%.
- Trainiere einen Logistic Regression Classifier auf den train Daten.
- Speichere Metriken auf den Validation Daten.
- Zurück zu Step 2 mit der nächsten GO.
Wichtig zu beachten: Die Trainings- und Validierungsdaten sind hier natürlich drastisch reduziert im Vergleich zum neuronalen Netz. Daher sind die Metriken nicht unbedingt zu vergleichen.
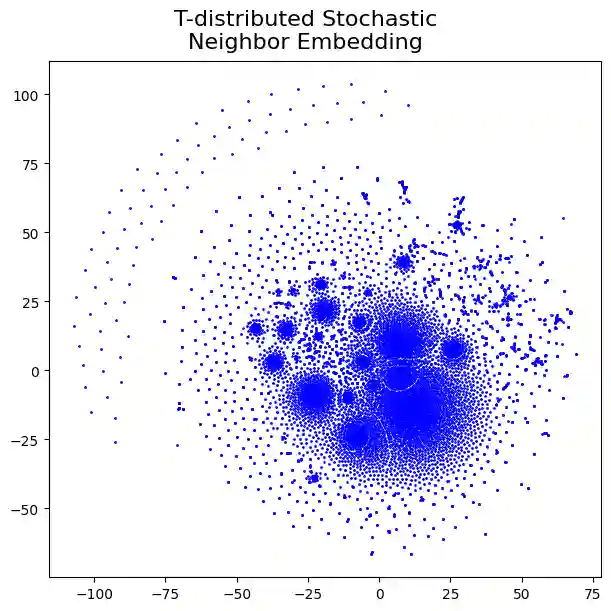
In dem Histogramm sieht man, wie viele Classifier (=Klassen) welche F1/Precision/Recall Werte haben.
Man erkennt, dass die einzelnen Classifier sehr gut performen und F1 Scores von über 0,7 oft vorkommen. Wie der F1 Score dann auf dem gesamten Validierungs-Datensatz ist, kann allerdings nicht direkt abgelesen werden.
Nun kommen wir zur Problematik dieses Ansatzes: Im Test-Datensatz (nicht Validierung!) der CAFA Challenge sind ca. 140.000 Sequenzen, deren GO Terms man vorhersagen soll. Mit der 1-vs-Rest Methode heißt das 140.000 mal jeden einzelnen Classifier aufrufen, also:
- Bei den 21.000 BPO Terms: 2,9 Mrd.
- Bei den 7.000 MFO Terms: 1 Mrd.
- Bei den 3.000 CCO Terms: 400 Mio.
In der Praxis würde man natürlich für jeden Classifier einen ganzen Batch an Sequenzen übergeben, das macht die Sache aber nicht unbedingt besser.
Auf meinem PC hätte das Ganze um die 20h gedauert, daher habe ich diesen Versuch vorerst abgebrochen.
Domains, Motifs und Superfamilies: Das InterPro Model
Die Performance mit den k-Mers war meiner Meinung nach ausgeschöpft, daher muss es noch weitere Daten geben, die die Funktion des Proteins beschreiben. Einfach nur zu wissen, welche Aminosäuren-Tripletts wie oft vorkommen, hat ja keinen direkten Bezug zur Funktion.
Eine Funktion eines Proteins ist eher durch das Zusammenspiel längerer Sequenzen innerhalb des Proteins bestimmt. Z.B. kann ein Transmembran-Protein nur funktionieren, wenn eine Substanz erkannt wird und durch Ladungsverschiebungen die Struktur verändert wird. Dafür sind lange Aminosäureketten notwendig, die genau das machen können.
Genau solche längeren zusammenhängenden funktionalen Gruppen werden in vielen Datenbanken gespeichert. Dazu gehören PANTHER, Prosite, CCD, …
InterPro ist ein großer Zusammenschluss all dieser Datenbanken. Zusätzlich werden auch Millionen von Proteinen klassifiziert und zu Domains, Sites, Families, … zugeordnet.
Eine Family ist dabei eine übergeordnete Gruppe von Proteinen, eine Domain ist eine funktionelle Gruppe in einem Protein. Sites sind dagegen kurze Sequenzen, die eine bestimmte Bindung ermöglichen.
Kurz gesagt: In der InterPro Datenbank stecken unglaublich viele Informationen zur Funktion eines Proteins.
Um eine solche Datenbank sinnvoll nutzen zu können, gibt es ein spezielles Tool. InterProScan kann genutzt werden, um einen beliebige Proteinsequenz gegen die gesamte Datenbank abzugleichen. D.h. wenn in der Sequenz eine bestimmte Domain vorkommt, wird diese erkannt und zugeordnet. Genauso für alle anderen Typen.
Beim Ausführen des Scans musste ich zwei Dinge beachten:
- InterProScan läuft nur auf Linux, d.h. Ich habe einen Docker Container genutzt, um die Skripte auszuführen, und die Trainings-/Testsequenzen via Volumes kopiert.
- Das Abgleichen der Sequenzen wird parallel ausgeführt und nutzt teilweise die komplette CPU und RAM. Bei 140.000 Proteinen kommt man da schnell an die Hardware Grenzen, also habe ich Test und Trainingsdaten in 6 Batches gesplittet.
Das Model sollte also als Input die InterPro Klassen und als Output die GO Klassen haben. Für beide wurde das One-Hot-Encoding angewendet, also wie oben schon beschrieben ein Vektor der 1 ist wenn eine Klasse vorhanden ist und 0 wenn nicht:
Alle Superfamilies, Domains und Sites in der Interpro Datenbank entsprechen ca. 38.000 Klassen. Der Output sind in diesem Fall die 2055 CCO (cellular component) Klassen.
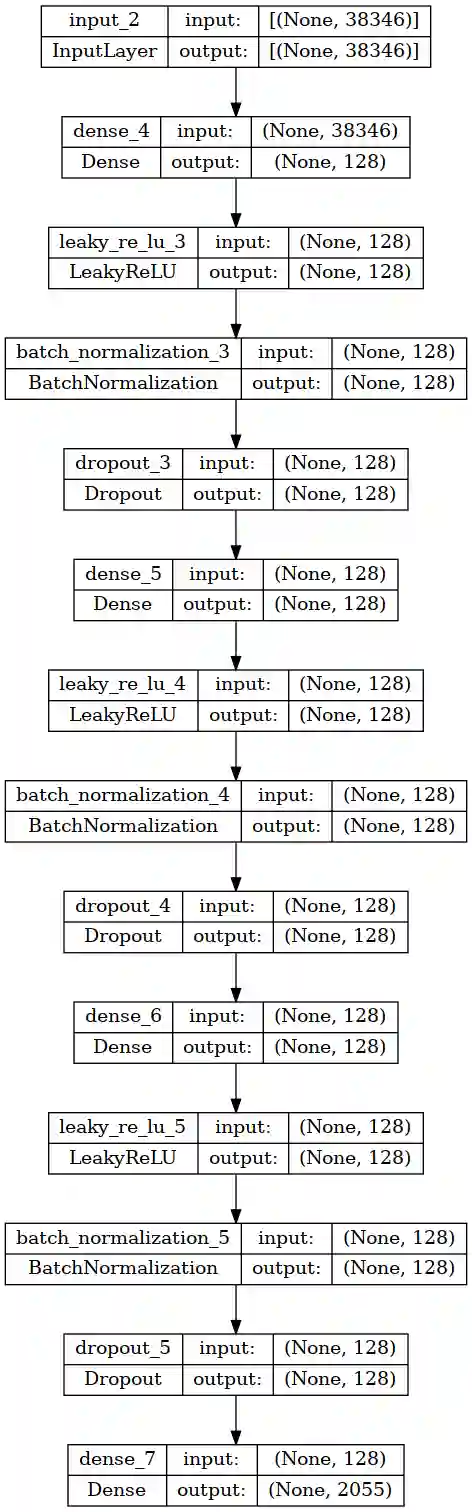
Vielleicht ist es dem ein oder anderen aufgefallen, dass ich vorher 2099 Klassen CCO Klassen hatte. Die Reduktion kommt von einem Filterprozess den ich zusätzlich noch eingebaut habe:
Bei dem 1-vs-Rest-Ansatz gab es ein paar Classifier, die überhaupt nicht gefittet werden konnten. Das lag daran, dass es im gesamten Datensatz nur eine einzige Sequenz gab. Also wurde hier noch ein Preprocessing Step angewendet: Pro GO-Klasse muss es mindestens 5 Sequenzen im Datensatz geben. Bei weniger ist es so gut wie unmöglich einen Classifier zu fitten der sinnvolle Unterscheidungen macht.
Die Architektur ist mit 4 hidden layers (128 Neurons), Batch Normalization, Leaky ReLu und Dropout ein gewöhnlicher Classifier mit Regularisierung.
Das Ergebnis: F1 = 0,22 auf dem Validierungs-Datensatz. Und damit leider keine Verbesserung zum k-Mer Model.
Es muss also ein neuer Ansatz her, der noch mehr Informationen enthält.
Mehr als nur ein Input für die Protein-KI?
Insgesamt hatten wir jetzt schon drei verschiedene Inputs: Die Aminosäure-Sequenz selbst, k-Mers und InterPro Klassen.
Doch was wäre, wenn man alle Inputs in einem großen Model kombinieren könnte? Kann man: Mit einem Multimodal Ansatz lassen sich die verschiedensten Features auf einen Feature Vektor reduzieren. Diese Feature Vektoren werden dann kombiniert und zum Schluss folgt der übliche Classifier.
Die Aminosäure-Sequenzen habe ich hier nicht weiter benutzt, da das bisher die schlechtesten Ergebnisse geliefert hat. Stattdessen wird als dritter Input folgendes benutzt:
Bisher waren die Input Features eher auf die mikroskopischen Eigenschaften ausgelegt. Also welche Aminosäuren wie oft vorkommen und was für Sequenzen es im Protein gibt. Was noch fehlt sind die makroskopischen Eigenschaften vom Protein als Ganzes: Molare Masse, Gesamtladung, Anteil an Sekundärstrukturen, Aminosäurenanteile, Aromatizität, etc.
All diese Eigenschaften können mit Biopythons ProteinAnalysis berechnet werden. Insgesamt ergibt das 53 neue Inputs für jedes Protein.
Und das Gesamtmodel sieht dann folgendermaßen aus:
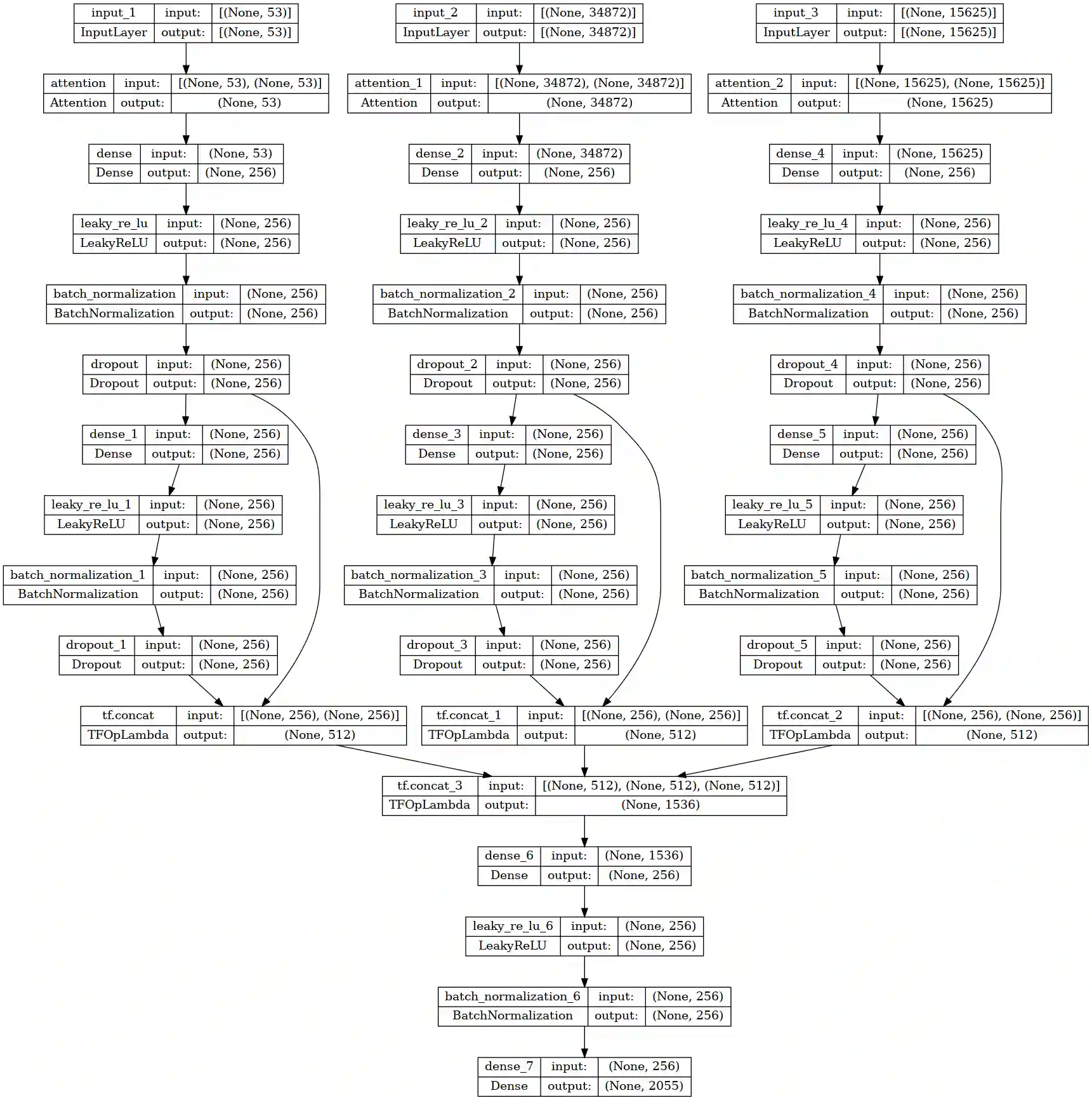
Von links nach rechts: Der erste Input sind die neue 53 Physiochemical (PC) Properties. Nummer 2 sind die InterPro Klasen und Input 3 sind die k-Mers.
Alle drei durchlaufen ein Attention Layer, gefolgt von der üblichen Dense+LeakyReLu+BatchNorm+Dropout Kombination. Das wiederholt sich nochmal und das Zwischenergebnis wird mittels Skip Connection zu einem Vektor der Größe 512 zusammengefügt.
Das Attention Layer zu Beginn ist sinnvoll, da viele der Input-Klassen möglicherweise komplett unwichtig für die Klassifizierung sind. So kann das neuronale Netz selbst entscheiden, welche Inputs auch wirklich genutzt werden.
Zuletzt werden noch alle drei Feature Vektoren zu einem 1536 großen Vektor zusammengefügt und mit einem “Classifier-Head” zu den 2055 CCO-Klassen transformiert.
Bevor wir zum Ergebnis kommen hier ein Diagramm zum Trainingsverlauf der zuvor beschriebenen Models:
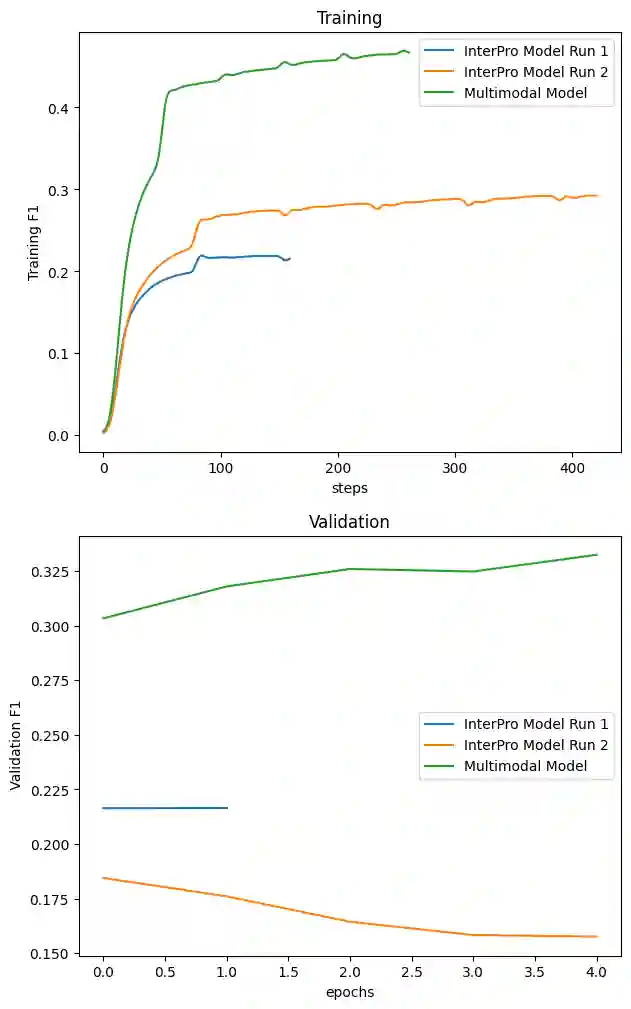
Hier werden die F1 Scores in Training und Validation für verschiedene Models und Runs gezeigt (Die Kurven sind leicht geglättet). Das InterPro Model aus Run 1 konnte auf die besagten F1_val = 0,22 trainiert werden. In Run 2 zeigt sich dann deutliches Overfitting, d.h. die relevanten Sequenzen aus dem Trainingsdatensatz werden einfach auswendig gelernt vom neuronalen Netz.
Der Multimodal Ansatz bringt einen ordentlich Sprung nach oben. Im Training werden F1 Werte von über 0,4 erreicht und im Validierungs Datensatz ergibt das F1 = 0,35.
Damit bringt das Multimodal Model ein deutlich größeres Potential zum Klassifizieren der GOs.
Ab hier lohnt sich der Blick in die Kaggle Community dieser Challenge und vor allem auch aufs Scoreboard. Denn eines wird deutlich: Keines der Top Submissions nutzt nicht mindestens eine Form von Embedding…
Proteine in Zahlen übersetzen: Embeddings
Embeddings hatte ich schon ganz zu Beginn im RNN genutzt. Allerdings ohne großen Erfolg. Der Unterschied zu den Top Submissions ist, dass hier vortrainierte Embeddings verwendet werden.
Das sind dann sehr große NLP Models die speziell auf Proteinen und Aminosäuren trainiert wurden (z.B. ProtBert) und so jedem beliebigen Protein einen Embedding Vektor zuordnen. Dieser Vektor beschreibt dieses Protein genau so, dass ein neuronales Netz bestmöglich damit arbeiten kann.
Von verschiedenen Nutzern auf Kaggle wurden die T5 und ESM2 Embeddings zur Verfügung gestellt. Für jedes Protein gab es damit einen 1024 (T5) oder 2560 (ESM2) langen Vektor, mit dem das Training eines Classifiers deutlich einfacher ist als mit den Sequenzen selbst.
Viele öffentliche Notebooks nutzen einfach nur die T5 Emeddings mit einem simplen neuronalen Netz mit einem hidden layer und erreichen schon F1 Werte von über 0,4.
Zum Testen habe ich die Inputs Stück für Stück in den Multi Modal Ansatz eingefügt, angefangen mit den ESM2 Embeddings:
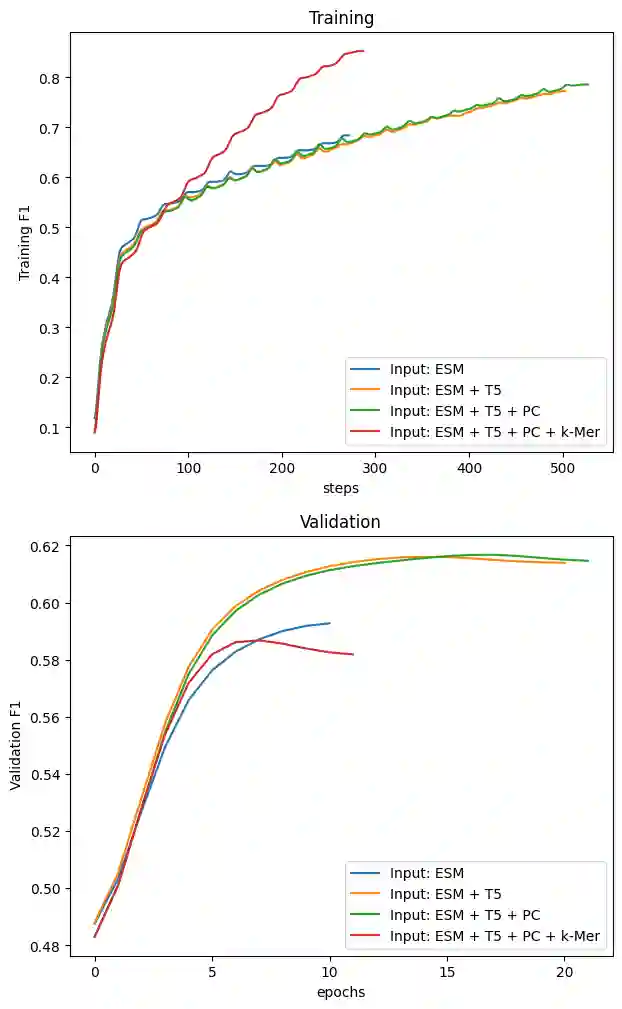
Man erkennt sofort, wie viel Informationen in den Embeddings stecken: Auf den Trainingsdaten ein F1 Wert von über 0,6 und das sogar nur mit ESM Input.
Bei dem Model mit 2 Inputs (ESM + T5) verändert sich das Training nur wenig, aber bei der Validierung ergibt sich nochmal ein deutlicher Zuwachs. Und mit 3 Inputs (zusätzlich noch die Physicochemical Properties) kann man noch eine kleine Verbesserung in der Validierung erkennen.
Problematisch wird es erst bei 4 Inputs. Die k-Mer machen das Modell und dessen Parameter viel komplexer und damit neigt es eher zum Overfitting. Die beiden roten Kurven zeigen lehrbuchmäßig wie Overfitting aussieht: Im Training steigen die Metriken fast bis zur Perfektion und in der Validierung fällt es ab.
Die Trainings oben wurden für MFO (molecular function) durchgeführt und das beste Ergebnis waren F1 = 0,62 auf dem Validierungs Datensatz. Das ist eine Verdopplung zu dem Model ohne Embeddings!
Hier zeigt sich: Die wahre Power der großen NLP Modelle auf denen auch ChatGPT, Bard, etc. basieren liegt in den Embeddings. Also die Fähigkeit, Wörter und deren Kontext in Zahlen und Vektoren umzuwandeln, ist das, worauf es ankommt.
Mein finales Modell sieht wie folgt aus:
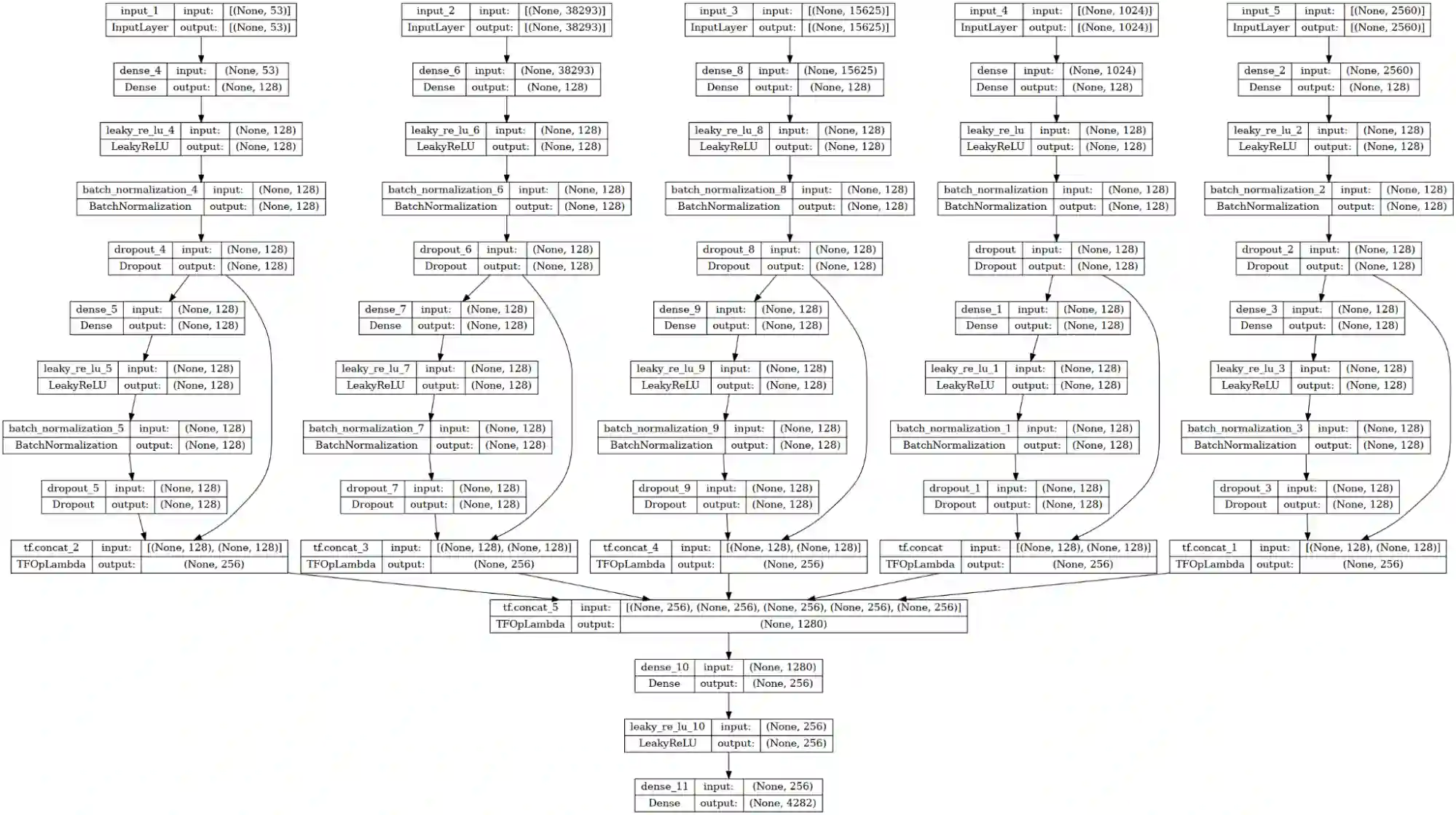
Es werden 5 Inputs verwendet (v.l.n.r.):
- PC Properties
- InterPro Klassen
- k-Mers
- T5 Embeddings
- ESM2 Embeddings
Der Output sind die (gefilterten) 4282 MFO-Klassen. Für CCO und BPO wurden dementsprechend die gleichen Modelle nur mit anderem Output erstellt und trainiert.
Da wir bei 4 Inputs schon Overfitting beobachtet haben, gab es noch Anpassungen: Die Attention Layer haben bei einem kurzen Test keine wirkliche Verbesserung gebracht, daher wurden diese entfernt. Die hidden layer haben nur noch 128 statt 256 Neurons und es gibt ein weiteres Dropout Layer. Außerdem habe ich noch die L2 Regularisierung erhöht.
Das Training wurde mit Early Stopping Kriterium durchgeführt, d.h. Die Modelle wurden nur so lange trainiert, bis es keine Verbesserung bei der Validierung mehr gab.
Die Ergebnisse auf dem Validierungs Datensatz:
- MFO: F1 = 0,649
- CCO: F1 = 0,610
- BPO: F1 = 0,456
Und damit wurden sehr gute F1 Werte erreicht, die jetzt natürlich noch auf den Testdaten angewendet werden müssen. Also alle Testdaten durch alle 3 Modelle durchschicken und die Submission als .csv Datei auf Kaggle hochladen.
Unter den 1629 Teilnehmern liege ich mit dem 382. Platz in den oberen 25%. Da das Projekt nebenher als Experiment und Erfahrung im Bioinformatik Bereich dienen sollte, bin ich damit mehr als zufrieden!
Danke fürs Lesen und bei Fragen, kann man mich gerne per Mail oder Kontaktformular kontaktieren.
Der Code ist auch auf GitHub verfügbar.